
Gaucher
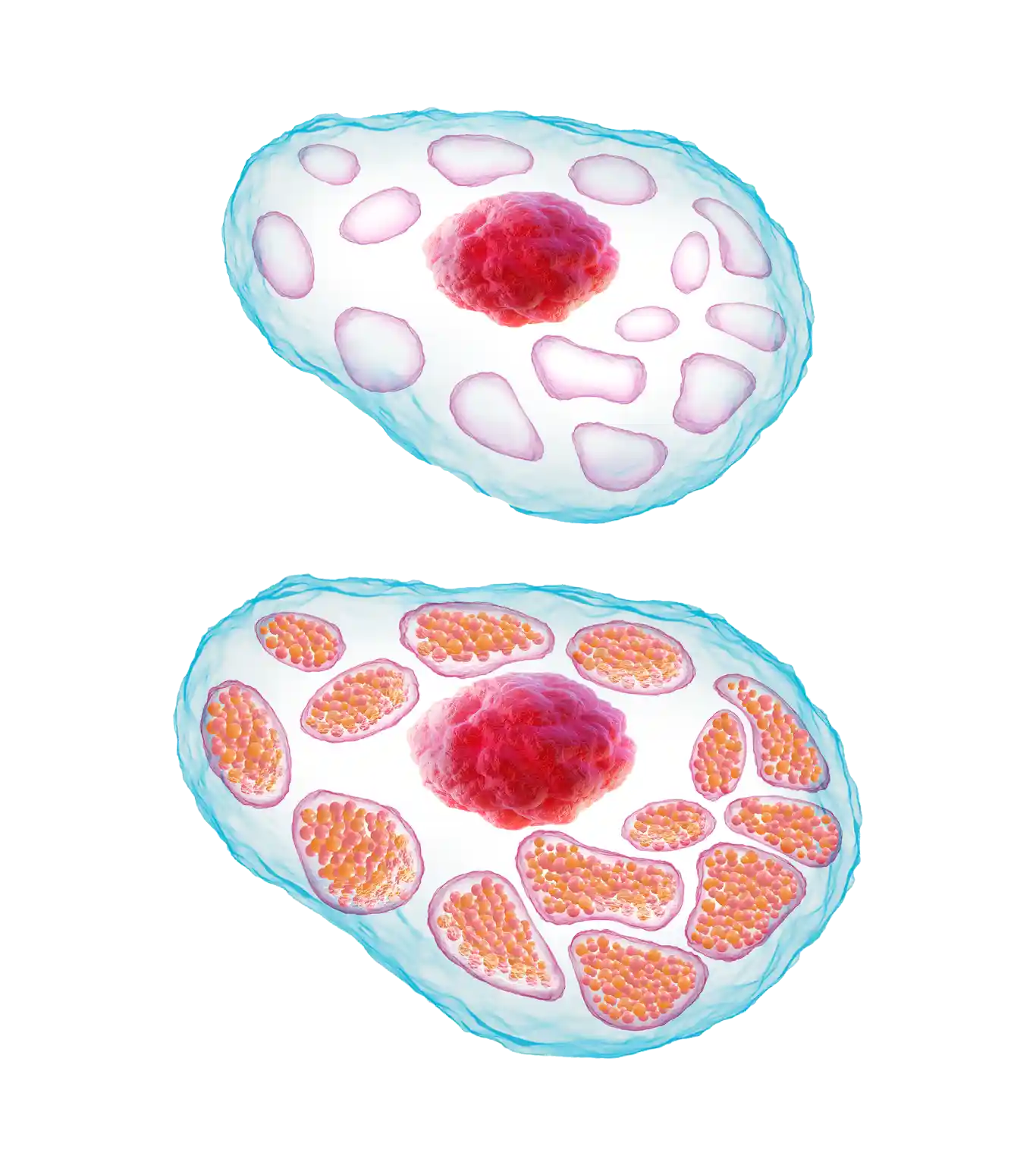 Healthy Cell
Gaucher Cell
Healthy Cell
Gaucher Cell

Prevalence, Genetics, and Inheritance
Both sexes can be affected by GD1, with males and females sharing an equal risk.3 GD1 is a pan-ethnic condition, although it does have a higher prevalence in people of Ashkenazi Jewish ancestry.5
GD1 affects:
- 1–9 in 100,000 within the overall population12
- ~1 in 600 within the Ashkenazi Jewish population3
- ~1 in 17 within the Ashkenazi Jewish community that are carriers13
- an estimated 6,000 individuals in the United States3,14
- GD1 is a lysosomal storage disorder and is caused by mutations of the GBA1 gene that controls the production of the glucocerebrosidase enzyme.14-16
- To date, more than 450 mutations in GBA1 have been identified.17
- The type of mutation may to some extent determine the type of GD, but there are other factors that affect the phenotypic expression. This has been demonstrated by cases in which symptom type, severity, and disease course have varied among siblings, even identical twins.6,18
GD1 is genetic and has an autosomal recessive inheritance pattern.3 If both parents are carriers, there is a 50% chance of the child being a carrier, a 25% chance of them having Gaucher disease, and a 25% chance that they will not be affected.3
Pedigree Diagram Illustrating the Autosomal Recessive Inheritance Pattern of Gaucher Disease

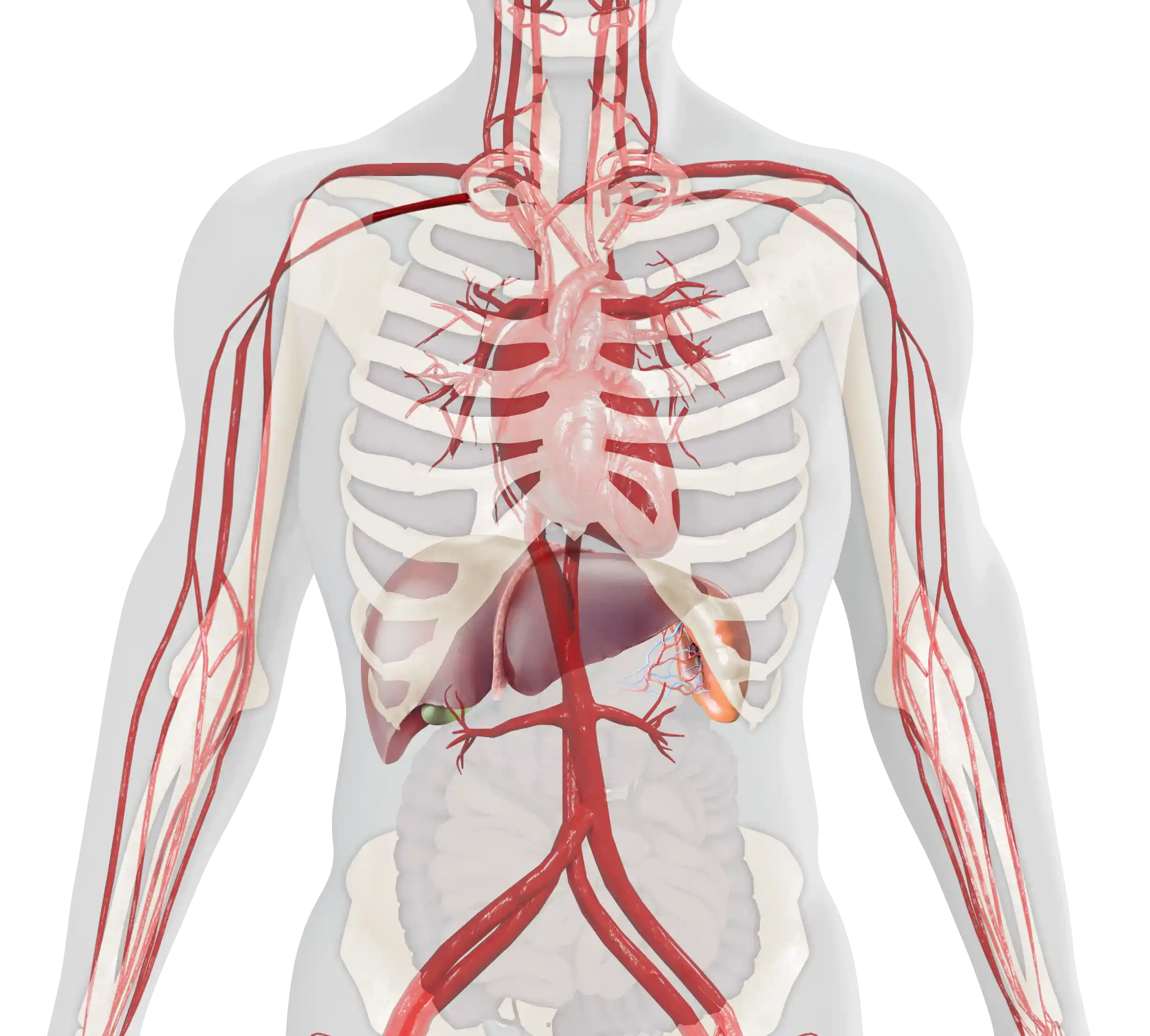
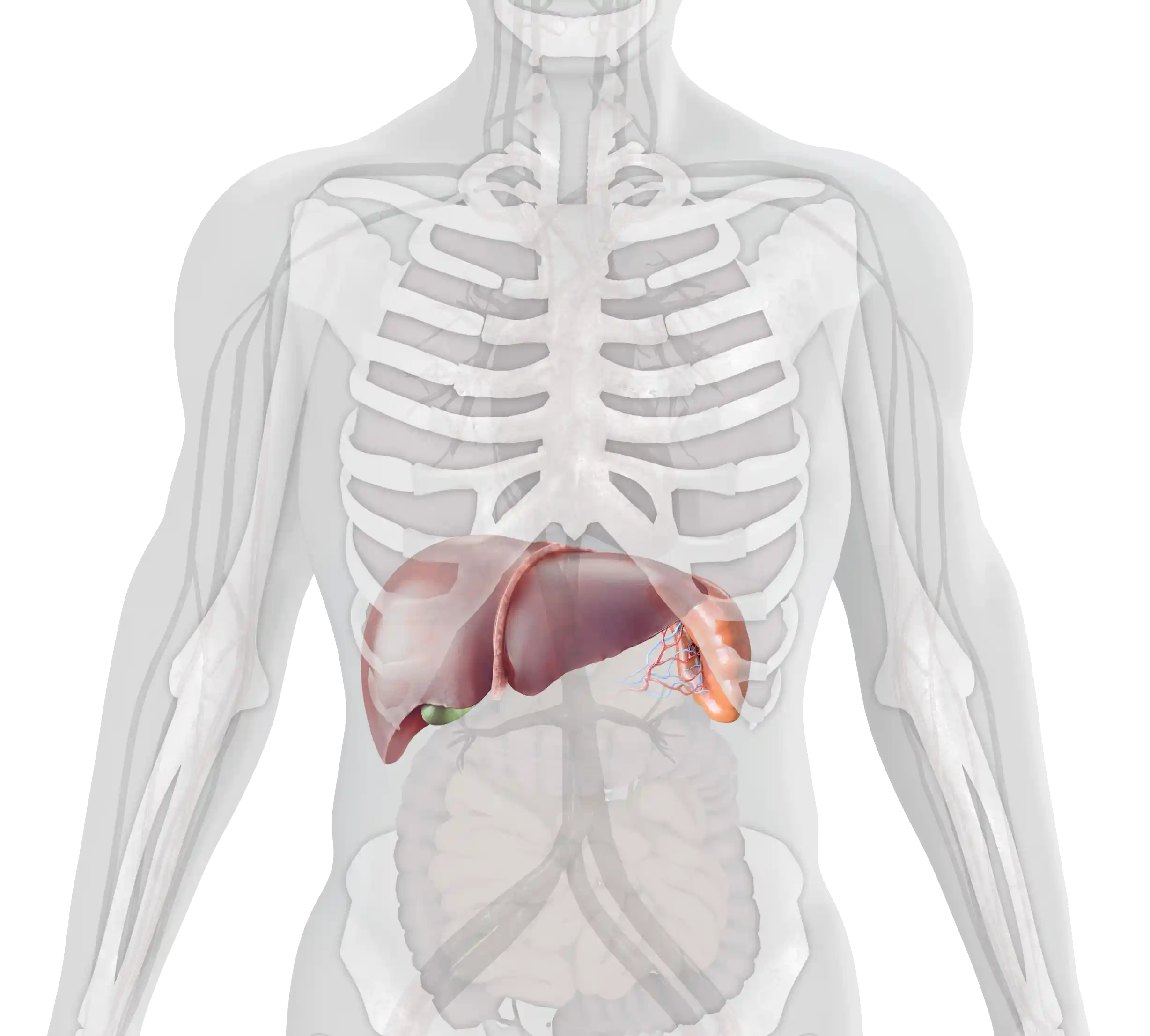
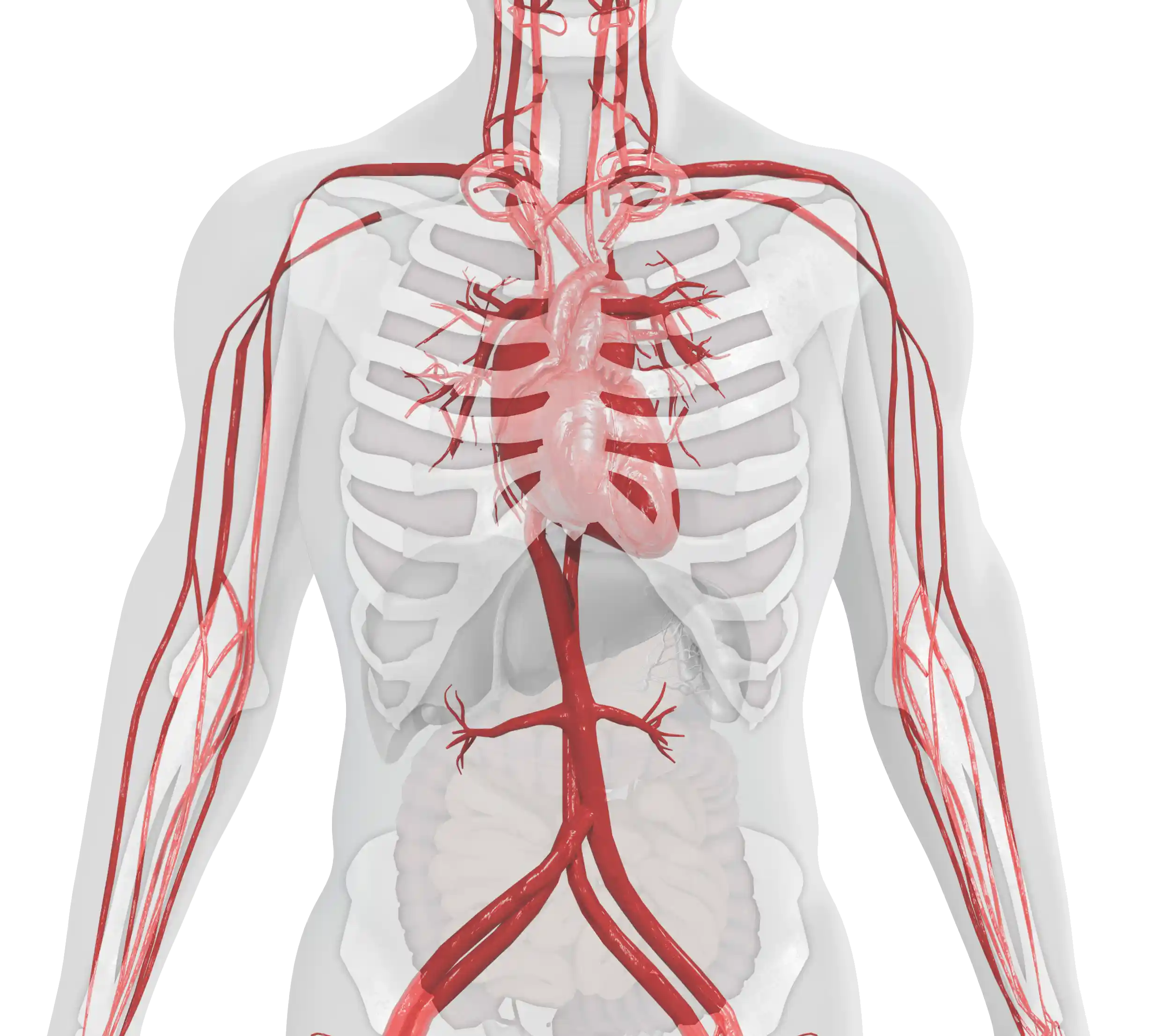
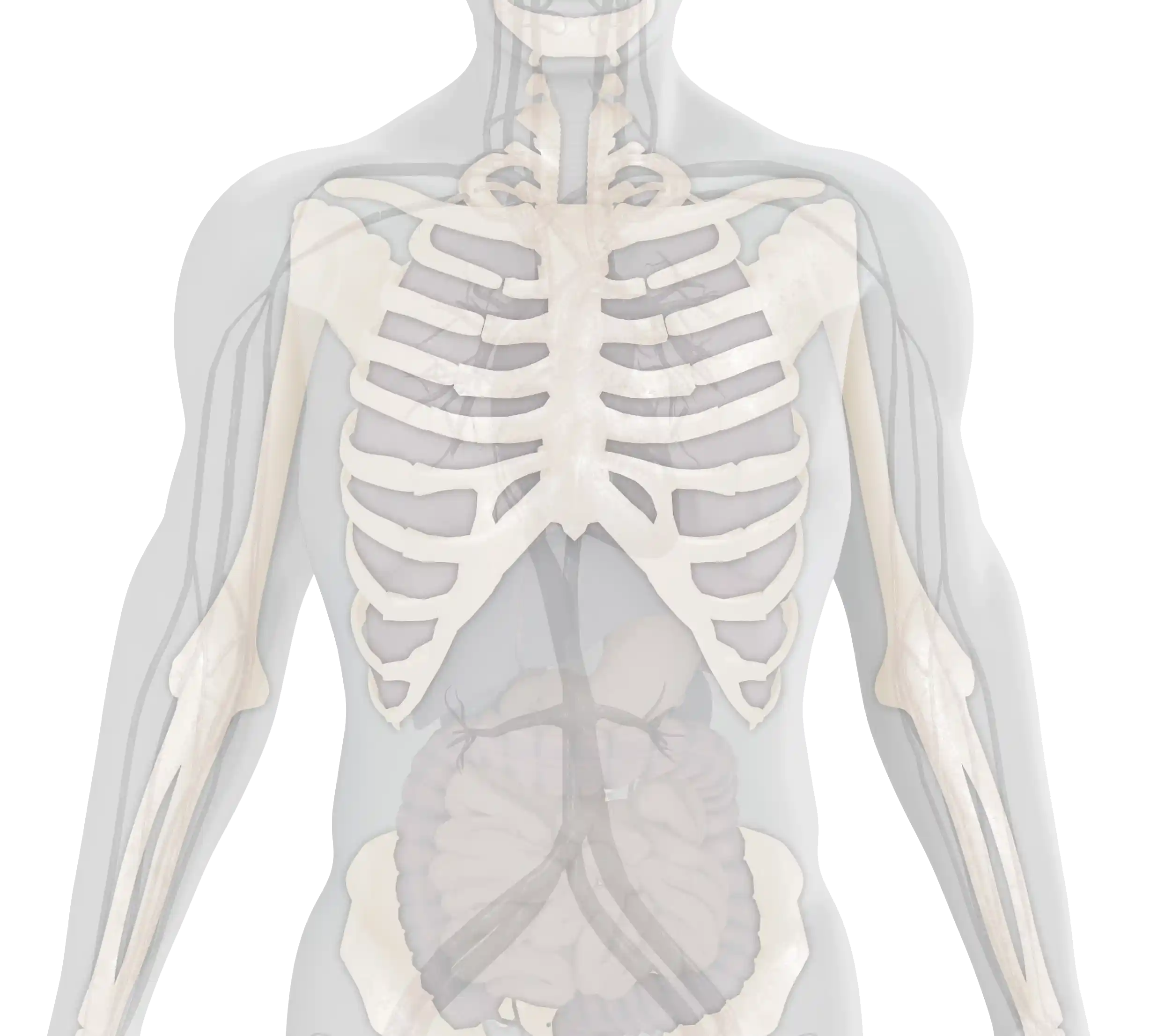
The age of onset and the severity of symptoms of type 1 Gaucher disease (GD1) can vary considerably.1,2 The main clinical manifestations include:
Visceral

Hematological
Skeletal
This summarizes the key clinical manifestations of GD1; however, a GD1 diagnosis and the resulting symptoms patients experience can also lead to further, long-term impacts on patients that require ongoing Monitoring and Management. Visit the Further Impact section to learn more.



Testing
The β-glucosidase enzyme assay measures the levels of the enzyme glucocerebrosidase.5 A GD diagnosis is confirmed by establishing glucocerebrosidase enzyme activity in mononuclear cells, leukocytes, or cultured fibroblasts. These are obtained by either a skin biopsy or dried blood spot test.6–8 Low levels of this enzyme, less than 15% of mean normal activity, indicate a diagnosis of GD.6,9 Enzyme activity, however, does not predict disease severity.1,6
Genetic testing requires either a blood or saliva sample, used to extract the patient’s DNA.10 Genetic sequencing can then be used to detect specific mutations in the GBA1 gene that result in GD.11,12
This algorithm* provides information to assist in the differential diagnosis of GD1 in patients with unexplained splenomegaly and/or thrombocytopenia.
This algorithm is not intended to be a diagnostic tool. It does not replace the need for a complete evaluation of the patient by a healthcare professional.
In cases of splenomegaly and/or thrombocytopenia it is important to include GD1 in differential diagnoses in order to avoid potentially harmful splenectomy or irreversible complications.7,15
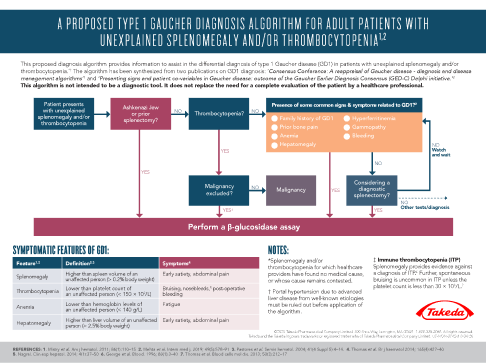

*This algorithm has been synthesized from two publications on type 1 Gaucher disease diagnosis: ‘Consensus Conference: A reappraisal of Gaucher disease – diagnosis and disease management algorithms’ and ‘Presenting signs and patient covariables in Gaucher disease: outcome of the Gaucher Earlier Diagnosis Consensus (GED-C) Delphi initiative.’3,13
**This diagnostic algorithm for pediatric patients has been synthesized from two publications on type 1 Gaucher disease diagnosis: ‘The diagnosis and management of Gaucher disease in pediatric patients: Where do we go from here?’ and ‘Screening, patient identification, evaluation, and treatment in patients with Gaucher disease: Results from a Delphi consensus.’6,14








Downloadable Materials
The following resources might be useful in your clinical practice to provide further information on managing patients with type 1 Gaucher disease (GD1):

Use this algorithm to learn more about the common features of GD1 and help diagnose adult patients with unexplained splenomegaly and/or thrombocytopenia.

Use this algorithm to learn more about the common features of GD1 specific to children and adolescents and help diagnose pediatric patients with unexplained splenomegaly with thrombocytopenia and/or anemia.

This flyer provides guidelines for the clinical monitoring of adult patients diagnosed with type 1 Gaucher disease and highlights the benefits of ongoing monitoring.

This flyer provides guidelines for the clinical monitoring of pediatric patients diagnosed with type 1 Gaucher disease and highlights the benefits of ongoing monitoring.

This brochure outlines three steps toward a better understanding of type 1 Gaucher disease, with the intention of spreading awareness of GD1.
There are also resources available that you can share with your patients to help them on their journey with GD1:

This caregiver guide, for anyone caring for a patient with GD1, is available for download below in both English and Spanish.

Other Resources
There are a number of organizations that specialize in raising GD1 and wider rare-disease awareness and building communities for patients living with these diseases. We encourage you to utilize the information and resources offered by these organizations and to make your patients aware of the additional support that they offer.
The NGF supports physicians in their efforts to better understand the diagnosis and treatment of Gaucher disease. Information and resources can be found on their website. Resources for your patients and their families are also provided.
The GCA is a patient organization that aims to support the patient community. Resources and opportunities to connect with other patients are available for your patients.
NORD provides resources for clinicians about specific rare disorders, including type 1 Gaucher disease, to facilitate the timely diagnosis and treatment of their patients. NORD also provides programs and services to support your patients.
CheckRare is the only publishing and learning platform dedicated exclusively to rare diseases and cancers. Visit to find original content from a range of perspectives, including patients and healthcare professionals, as well as industry and regulatory bodies.


SITEMAP
©2024 Takeda Pharmaceuticals U.S.A., Inc., 500 Kendall Street, Cambridge, MA 02142.
1‑877‑TAKEDA‑7 (1‑877‑825‑3327).
All rights reserved. Takeda and ![]() are registered trademarks of Takeda Pharmaceutical Company Limited.
US‑NON‑8044v2.0 12/24
are registered trademarks of Takeda Pharmaceutical Company Limited.
US‑NON‑8044v2.0 12/24
All images used on this website are stock photography images and do not portray actual patients.

Hematologist

Geneticist
Healthy Cell
Gaucher Cell